| Hemoglobinopatias, Estudo | ||||||||||||||||
| SINONÍMIA: PESQUISA DE TALASSEMIAS MATERIAL: SANGUE TOTAL (EDTA) VOLUME MÍNIMO: 3 ml DIETA: JEJUM 4 HORAS CONSERVAÇÃO PARA ENVIO: CAIXAS DE ISOPOR COM GELO RECICLÁVEL (NÃO CONGELAR) ATÉ 5 DIAS APÓS A COLETA
MÉTODO: Eletroforese alcalina, ácida e pesquisa citológica INTERPRETAÇÃO: DIAGNÓSTICO DE TALASSEMIAS E/OU HEMOGLOBINOPATIAS. |
||||||||||||||||
TALASSEMIA
As talassemias representam um conjunto de sintomas caracterizados por anemia, fraqueza, cansaço, dores nas pernas, palidez, icterícia, e que dependendo da gravidade da doença, ocorrem alterações esqueléticas, hepatomegalia, esplenomegalia, cálculos biliares, infecções, insuficiência cardíaca e disfunções hormonais. Cientificamente, as talassemias foram descritas pela primeira vez por Thomas B. Cooley em 1925, que relatou a doença em quatro crianças que tinham anemia e esplenomegalia, aumento do fígado, palidez, todos com aspectos faciais mongolóides, e alterações ósseas do crânio e dos ossos faciais.
No exame de esfregaço sangüíneo, Cooley descreveu acentuado grau de anemia com presença de muitos eritroblastos. Devido à excelência do artigo e da clareza com que foi descrito, por muitos anos essa forma de talassemia grave foi reconhecida e citada como anemia de Cooley, e que hoje corresponde à talassemia beta maior. Historicamente, é possível que a talassemia tenha se originado há mais de 4 mil anos, pois há referência da doença feita por Hipócrates, bem como achados em múmias de crianças dos períodos pré-histórico e histórico, com alterações ósseas no crânio, muito parecidos com os da talassemia beta maior.
Os gregos certamente já conheciam a talassemia como uma entidade mórbida, distinguida em certas regiões e ilhas devido aos casamentos consangüíneos. Por essa razão, é provável que daí tenha se originado o nome talassemia, onde thalassa significa mar; e anaima que sugere "pessoas com falta de sangue (anêmicas) que vivem a beira-mar (ilhas)", surgindo daí o termo talassemia. Atualmente, a talassemia é uma das doenças mais estudada e mais conhecida no sentido científico, médico, social e demográfico.
Sob o ponto de vista clínico, as talassemias são classificadas em maior, intermediária, menor e mínima, conforme mostra a tabela 1. Por outro lado, as talassemias também têm sua classificação laboratorial conforme o tipo de globina que foi afetada, fato que as diferenciam em:
a. talassemia alfa: quando a síntese da globina alfa está deficiente, enquanto a de globina beta está normal;
b. talassemia beta: quando a síntese da globina beta está deficiente, enquanto a de globina alfa está normal.
O desequilíbrio entre as globinas alfa e beta causa a precipitação dentro do eritrócito da globina que foi sintetizada normalmente, pois esta não encontrou a sua correspondente para se juntar e formar a molécula da hemoglobina.
Desta forma, conclui-se que quanto maior for o bloqueio da síntese de uma das globinas, maior será o desequilíbrio, mais intenso serão as globinas precipitadas, e mais extensas serão as lesões nos eritrócitos – que serão retirados precocemente da circulação causando anemias hemolíticas de graus variados. Além das talassemias alfa e beta, há vários tipos de lesões moleculares que ocorrem nos cromossomos 16 (onde estão agrupados os genes da globina alfa) e 11 (onde estão agrupados os genes das globinas beta, delta e gama).
Assim, é possível ter situações de talassemias em que as globinas alfa e beta estão deficientes numa mesma pessoa: talassemia alfa/beta; situações em que as globinas beta e delta estão deficientes: beta/delta talassemia, etc. E também ocorrem as interações entre talassemias e hemoglobinas variantes, como são os casos da Hb S/tal. beta, Hb SS/tal. alfa, Hb C/tal. beta, etc.
ALFA TALASSEMIA
Os distúrbios genéticos que causam a talassemia alfa se devem às lesões nos agrupamentos de genes para síntese de globinas alfa que estão localizados no braço curto do cromossomo 16. Uma pessoa que não tem talassemia alfa apresenta dois genes alfa íntegros em cada um dos dois cromossomos 16 (um herdado do pai e outro da mãe), assim, há quatro genes alfa atuantes.
A talassemia alfa ocorre quando um, ou dois, ou três, ou os quatro genes alfa estão alterados nos seus processos biológicos de síntese de globinas alfa. Assim, é possível afirmar que os tipos de talassemias alfa se devem ao número de genes afetados. Fisiopatologicamente, as conseqüências clínicas e hematológicas de quem padece de talassemia alfa se devem à quantidade intra-eritrocitária de globina beta "livre".
Quanto maior a extensão da lesão de um gene alfa, ou de dois, três ou dos quatro genes alfa, o desequilíbrio em relação à globina beta será maior, fato que permite diferenciar as talassemias alfa em três grupos básicos: portador assintomático (talassemias alfa mínima e menor), doença de Hb H e hidropsia fetal.
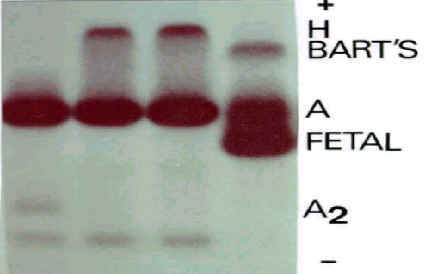
a. Quadro azul: a diminuição da síntese da globina alfa tem início na fase fetal, assim o desequilíbrio ocorre na combinação entre globinas alfa e gama. Na fase fetal, as globinas gama "sobram" e precipitam nos eritrócitos em forma de tetrâmeros de globinas gama (g4) e compõe moléculas instáveis de hemoglobinas – a Hb Bart’s . Essas hemoglobinas têm afinidade aumentada pelo oxigênio, não tem efeito Bohr, e não desempenham a interação entre grupos heme no processo de oxigenação molecular – ou seja, são hemoglobinas sem desempenho funcional. Ao nascer, esta mesma pessoa continua a sofrer as conseqüências do desequilíbrio causado pela deficiência da síntese de globina alfa, pois nesta fase a globina beta que substitui a globina gama, sofre a mesma conseqüência, uma vez que o tetrâmero formado somente de globinas beta (b4) constituem moléculas de hemoglobinas anormais, a Hb H , que se precipitam nos eritrócitos e são identificadas pela coloração citológica com azul de crezil brilhante.
 |
 |
BETA TALASSEMIA
A talassemia beta é uma das doenças mais bem estudadas e conhecida entre as de origem hereditária. A causa genética se deve a lesões provocadas por mutações de bases nitrogenadas nas regiões que controlam os genes do tipo beta, delta, gama e epsilon – este último com atividade somente no período embrionário. Esse agrupamento de genes, também conhecido genericamente como genes do tipo beta, estão localizados no braço curto do cromossomo 11. As lesões genéticas que ocorrem nos genes tipo beta podem afetá-los parcial ou integralmente, e podem também ser cumulativos, ou seja, podem a lterar a expressão só do gene beta, bem como dos genes beta e delta; beta, delta e gama; beta, delta, gama e epsilon, . Quando o bloqueio do gene é total, denomina-se por bo-talassemia, e quando o bloqueio é parcial, por b+ talassemia.
A forma de transmissão da talassemia beta é variável, e depende do tipo de lesão: total ou parcial. Uma pessoa sem talassemia beta tem dois genes beta (b/b), um dos genes beta proveniente do cromossomo 11 do pai e o outro da mãe. Além dos genes beta, há os genes delta, gama e epsilon, porém por praticidade o exemplo se concentrará apenas nas lesões dos genes que sintetizam a globina beta. Quando o bloqueio do gene beta é total e afeta apenas um dos dois genes da pessoa (b/-), o portador é caracterizado geneticamente como talassemia beta heterozigoto, ou clinicamente como talassemia beta menor.
Por outro lado, a lesão total de ambos os genes beta causa a talassemia beta homozigota ou talassemia beta maior (tabela 1). Nestas duas citações de bloqueio total do gene beta, pode-se deduzir que o primeiro caso é bo talassemia heterozigota, e o segundo se trata de bo talassemia homozigota. O mesmo procedimento de interpretação pode ser feito no bloqueio parcial da globina beta. Há situações de dupla heterozigose que causa clinicamente a talassemia beta maior, é o que ocorre quando há a herança por parte do pai de um cromossomo 11 com o gene beta bloqueado totalmente (bo) e do outro cromossomo 11 por parte da mãe com o gene beta bloqueado parcialmente (b+). Neste caso, a duplicidade de genes alterados bo/b+ causa a talassemia maior.
| Tabela 1 | ||||
| Talassemia Beta | ||||
| Maior | Intermédia | Menor | Mínima | |
Hemoglobina |
< 7 | 7 - 10 | 10 - 13 | > 13 |
Reticulócito (%) |
2 - 15 | 2 - 10 | 2 - 5 | 0,5 - 2 |
Eritroblasto |
+ + + | + + / + | - - - | - - - |
Eritrócitos Alterados |
+ + + | + + | + | + / - |
Icterícia |
+ + | + | - - - | - - - |
Esplenomegalia |
+ + + | + + | + / - | - - - |
Alterações Esqueléticas |
+ + + / + + | + + / + | - - - | - - - |
Necessidade de Transfusões |
+ + + | + / - | - - - | - - - |
 |